
با وجود اینکه مواردی از سرطان کلیه در جمعیت جوان جامعه مشاهده میشود، اما در سنین بالای 60 سال شایعتر است. شایعترین علامت اولیه، وجود خون در ادرار است. این سرطان، چنانچه درمرحله اولیه تشخیص داده شود، شانس خوبی برای درمان دارد. اما با پیشرفت سرطان (رشد و انتشار آن)، این شانس درمان کاهش مییابد. در هر صورت درمان، باعث کاهش سرعت پیشرفت بیماری میشود
بیماری کرونا ویروس2019 (Covid 19) که توسط سندرم شدید حاد تنفسی کرونا ویروس ۲ (SARS-CoV-2) ایجاد می شود، به سرعت به یک بیماری همه گیر جهانی تبدیل شده است. عوارض بالینی این بیماری بسیار متفاوت است.
اختلالات انعقاد خون و عوارض ترومبوتیک بعنوان مشکلات اساسی در بیماران مبتلا به کووید ۱۹ حاد می باشند که با میزان مرگ و میر بالایی همراه است. در اینجا عملکرد بالقوه و پیامدهای درمان با ضد انعقاد ها و درمان های ترومبولیتیکی در بیماران کووید ۱۹ را مورد بحث قرار خواهیم داد.

اگر شما تب دارید، سرفه می کنید یا علائم دیگر ویروس covid 19 را دارید ممکن است به بیماری کرونا مبتلا شده باشید.اکثر مردمی که به این بیماری مبتلا می شوند بیماری خفیف دارند و اگر مدتی در خانه استراحت کنند بهبود پیدا می کنند. در این مقاله راهکار های اساسی و اولیه برای مقابله و درمان بیماری کرونا بررسی می شود.

بیماری های مقاربتی (STD: Sexual transmitted Disease) از بیماریهای نسبتاً شایعی هستند که توسط میکروبها (موجودات ذره بینی) طی مقاربت جنسی واژنی، مقعدی و دهانی از فردی به فرد دیگر منتقل می شود. بیماری های مقاربتی باعث ایجاد علائم موضعی و عمومی در بدن میشوند. اگر نسبت به تشخیص بیماری های مقاربتی و درمان این نوع بیماری ها اقدام نشود مشکلات جدی مانند ناباروری، سرطان ها، کوری و غیره ایجاد خواهد شد.
انجام غربالگری های منظم می تواند باعث کاهش خطر انتقال بیماری های آمیزشی، کاهش ناباروری و سایر آسیب های جسمانی شود.
همان طور که می دانید ناراحتی قلبی، آسیب دیدن چشمها، عصبها و بافتهای دیگر از عوارض احتمالی دیابت محسوب میشود. اما در سالهای اخیر دانشمندان متوجه شدند دیابت احتمال ابتلا به آلزایمر یا همان فراموشی را افزایش می دهد. این ارتباط منطقی و کاملاً قابل توجیه است، بیماری دمانس عروقی پیآمد آسیب دیدن رگهای خونی مغز است، از طرف دیگردر دیابت رگهای بدن دچار سختی می شوند. تاکنون بسیاری از دانشمندان بر سر این موضوع به توافق رسیدهاند که اگر دیابت کنترل شود، احتمال ابتلا به آلزایمر به نصف تقلیل مییابد. در این مقاله ارتباط بین دیابت و آلزایمر توضیح داده می شود.

آزمایشگاه اریترون با هدف ارتقا سطح خدمات خود جهت صرفه جویی در وقت و هزینه مراجعین و همچنین جلوگیری از به خطر افتادن سلامتی مراجعین محترم، خدمات نوبت دهی، پذیرش و جوابدهی خود را به صورت غیر حضوری در شبکه های مجازی ارائه می دهد. برای پذیرش آنلاین آزمایشگاه اریترون، مراحل زیر را انجام دهید.

همه ما میدانیم که ریزش مو پدیده شایعی است که میتواند همه افراد را در گیر کند. اگر کسی در شبانه روز بیش از 100 تار مو از دست دهد باید به دنبال علت و درمان ریزش موی خود باشد چراکه در این شرایط تعادل بین رشد و ریزش مو به هم میخورد و به مرور زمان تراکم مو کاهش مییابد. دلایل ریزش مو بسیار متفاوت است. برای اینکه از ریزش مو جلوگیری کنید باید علت آن را بشناسید و هرچه سریعتر درمان مناسب را انتخاب کنید. در این متن راهکارهایی برای اینکه بدانید چگونه می توان از ریزش مو جلوگیری کرد، ارائه میشود.

سندرم کرست CREST یکی از انواع گروه ناهمگون اسکلرودرماها است. این سندرم نادر است و بیشتر در زنان ایجاد می شود. نام این بیماری مخفف 5 خصوصیت بالینی بارز این سندرم است که عبارت است از: کلسینوز Calcinosis، پدیده رینود Raynaud phenomenon، اختلال حرکتی مری Esophageal dysmotility، اسکلروداکتیلی ( تشکیل بافت لیفی و ضخیم) Sclerodactyly، تلانژکتازی (گشاد شدن مویرگها) Telangiectasia
علائم بیماری در افراد از خفیف تا شدید و کشنده دیده می شود. به دلیل اینکه درمان خاصی برای این بیماری وجود ندارد، بیشتر درمان ها تسکین دهنده علائم بیماری هستند. بنابراین تشخیص سندرم کرست اهمیت دارد. در این مقاله درباره این سندرم و روش های تشخیص آن مطالبی ارائه می گردد.

ویروس پاپیلومای انسانی دارای بـیش از 200 نوع مـی باشد که 40 نوع از آنها از طـریـق جنسـی و تمـاس پـوسـت با پوسـت می تواند منـتقـل شود. اکثر زنانی که از لحاظ جنسی فعال هستند، در معرض آلودگی با این ویروس قرار دارند. بعضی از انواع پر خطر این ویروس ها (14نوع) ممکن است باعث ایجاد سرطان دهانه رحم شوند. در این مقاله که در مجله هنر تشخیص آزمایشگاه اریترون هم منتشر شد، شیوهنامه جدید غربالگری و پیگیری ویروس hpv در مردان و زنان معرفی می شود.

ممکن است احساس کنید که نیازی به چکاپ بدن نیست اما ازنظر پزشکان و متخصصان، بررسی سالیانه بدن مهمتر از آن چیزی است که فکر می کنید. انجمن دیابت آمریکا پیشنهاد میکند که در ۶۳ درصد از موارد، حملات قلبی را میتوان با چکاپ بدن و استراتژیهای پیشگیرانه مناسب مهار کرد. چکاپ سالیانه به چه معناست؟ چه کسی نیاز به معاینه پزشکی دارد؟ چرا باید این کار را بکنی؟ در این مقاله به سؤالات و مسائل مربوط به چکاپ کامل بدن میپردازیم.
امروزه با توجه به شیوع بیماری کرونا در کشور و استفاده از انواع واکسنها، آزمایشهای سرولوژیک جهت رفع نیازهای تشخیصی موردتوجه قرارگرفته و توسعهیافتهاند. واکنشهای متقاطع، جهش و تنوع کرونا ویروسها و زمان مناسب سنجش آنتیبادیها از چالشهای آزمایشهای سرولوژیک کرونا است. بنابراین شناخت دقیق ویروس و انتخاب آزمایش آنتی بادی مناسب از اهمیت زیادی برخوردار است.

انتقال خون یکی از روشهای درمان و رهانیدن انسانها از خطر مرگ و میر ناشی از حوادث و بیماریه است که می تواند منجر به عوارض ناخواسته ای در گیرندگان خون شود؛ یکی از این عوارض، عفونتهای میکروبی قابل انتقال از طریق تزریق خون است. عفونتهای میکروبی شامل طیف وسیعی از میکروارگانیسمها مانند: ویروسها، قارچها، انگل ها و باکتری ها است. اولین مورد عارضه باکتریایی ناشی از تزریق خون انسان در ایالات متحده در سال 1941 گزارش شده است

یائسگی، دورانی در زندگی زنان است که قاعدگیهای ماهانه او حداقل 12 ماه متوقف میشود و دیگر نمیتواند باردار شود. معمولاً یائسگی یک مرحله تدریجی است که در سنین 45 تا 55 سالگی شروع میشود. گاهی ممکن است یائسگی به دلایل بیماری، اقدامات پزشکی یا اختلالات هورمونی، زودتر شروع شود. بنابراین برای تشخیص یائسگی به انجام آزمایشهای تکمیلی، نیاز است.

نوجوانان اغلب به عنوان سالم ترین گروه سنی در نظر گرفته می شوند. با این حال، عادت هایی که در طول سال های نوجوانی شکل گرفته اند، احتمالاً تا بزرگسالی بر سلامت نوجوانان تأثیر می گذارند. به عنوان مثال، اگر یک نوجوان دارای اضافه وزن، با رعایت رژیم غذایی و ورزش وزن خود را کم نماید می تواند از دیابت و بیماری های قلبی در سال های بعدی جلوگیری کند.
برای نوجوانان، همه معاینات سالانه شامل بسیاری از تست های غربالگری آزمایشگاهی نمی باشد بلکه تأکید بر آماده سازی برای مسائل بهداشتی نوجوانان، مانند پیشگیری از تصادف و آسیب جسمی، بهداشت جنسی و جلوگیری از سوء مصرف مواد مخدر است. داروهای پیشگیرانه برای نوجوانان باید بر انتخاب سبک زندگی سالم که به محافظت در برابر بیماری هایی که در بزرگسالی اتفاق می افتند، کمک کند. در بخشهای زیر چند بیماری که نوجوانان 13 تا 18 سال باید برای آنها غربالگری گردند، بیان شده است.
یکی از مهمترین دلایل حوادث ترومبوتیک و سقط مکرر تغییر در سطح پروتئین های سیستم ضد انعقاد است. پروتئینهای ضد انعقادی شامل آنتی ترومبین، پروتئین C و S است. ضد انعقادها فعالیت فاکتورهای انعقادی را تنظیم و مانع از ایجاد لخته خون در بدن می شود. تغییر سطح فاکتورهای انعقادی با خطر ایجاد لخته خون (آمبولی) همراه است. با تشخیص و درمان به موقع می توان از بروز خطرات جدی پیشگیری کرد.
کووید ۱۹ توسط کروناویروس SARS-CoV-2 که به بافت عصبی تمایل دارد، ایجاد می شود علائم اختلال در بویایی و چشایی بعنوان تظاهرات اولیه عفونت کووید ۱۹ پذیرفته شده است. گزارشاتی از آنسفالیت، آنسفالوپاتی، نوروپاتی کرانیال، سندرم گیلن باره (GBS) و التهاب میوزیت/ رابدومیولیز در بیماران مبتلا به کووید ۱۹ وجود دارد. در این مقاله، بر روی تظاهرات عصبی عضلانی عفونت SARS-CoV-2 و مکانسیم درگیری بافت های عصبی عضلانی در کووید 19 متمرکز خواهیم شد.

برای مهار پایدار همه گیری جهانی ویروس کووید 19 نیاز به مصون شدن بیش از 65 درصد از افراد جامعه است؛ انجام واکسیناسیون سراسری بهترین راه برای مهار این ویروس می باشد. تاکنون شرکت های زیادی در سراسر جهان موفق به تولید واکسن شده اند. برای درک چگونگی عملکرد واکسن کووید 19، بهتر است ابتدا نحوه مبارزه بدن ما با بیماریها بررسی شود. در این مقاله درباره نحوه عملکرد واکسن و انواع واکسن کووید 19 مطالبی ارائه می گردد.

گنادوتروپین جفتی انسانی یا hCG ((The human chorionic gonadotropin هورمونی است که توسط جفت یک زن باردار تولید میشود. در اوایل بارداری، سطح hCG در خون افزایش مییابد و از طریق ادرار دفع میگردد. در آزمایش بارداری، میزان hCG موجود در خون یا ادرار را اندازهگیری کرده و حاملگی را تأیید یا رد میکند.

کودکان معمولاً به آزمایشهای غربالگری زیادی احتیاج ندارند مگر اینکه علائم بیماری خاصی را داشته باشد. بااینحال، انجام آزمایش های غربالگری جهت کمک به کودکان در ایجاد عادتهای سالم، مانند خوب غذا خوردن و تحرک، میتواند از مشکلات بهداشتی جدی و پرهزینه با افزایش سن جلوگیری کند.
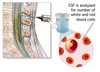
مایع مغزی نخاعی (CSF) مایعی شفاف و آبکی است که در اطراف مغز و نخاع جریان دارد و آنها را احاطه کرده و محافظت میکند. آنالیز CSF برای ارزیابی سطح یا غلظت مواد و سلولهای مختلف در CSF بهمنظور تشخیص شرایط بیماری بر مغز و نخاع (سیستم عصبی مرکزی) انجام میشود.
اریترون یک آزمایشگاه تخصصی است که از راه های مختلف میتوانید با آن در تماس باشید و پرسش ها و مشکلات خود را به آسانی با متخصصین ما در میان بگذارید.
آدرس: اصفهان، خیابان شیخ صدوق شمالی، خیابان شیخ مفید غربی
شماره تماس: 7-36631906- 031 2 -36633621 - 031
031-37134
شماره فکس: 89784728- 021 کد پستی : 76351-81647
ایمیل: [email protected]
شماره واتس آپ برای دریافت جواب آزمایش :
09138183947
برای هماهنگی جهت نمونه گیری در محل مورد نظر خود با شماره های زیر در ساعت مشخص تماس حاصل فرمایید:
آقای مهندس عزیزی 09131689270
تمام حقوق مطالب و محتویات این سایت متعلق به آزمایشگاه اریترون می باشد.